Thalasemia merupakan penyakit anemia hemolitik herediter yang diturunkan
secara resesif. Ditandai oleh defisiensi produksi globin pada hemoglobin.
dimana terjadi kerusakan sel darah merah di dalam pembuluh darah sehingga umur
eritrosit menjadi pendek (kurang dari 100 hari). Kerusakan tersebut karena
hemoglobin yang tidak normal (hemoglobinopatia)
Tanda Dan Gejala Klien Dengan Thalasemia
Klasifikasi Thalasemia
Secara molekuler talasemia dibedakan atas :
Klasifikasi Thalasemia
Secara molekuler talasemia dibedakan atas :
- Thalasemia a (gangguan pembentukan rantai a)
- Thalasemia b (gangguan p[embentukan rantai b)
- Thalasemia b-d (gangguan pembentukan rantai b dan d yang
letak gen nya diduga berdekatan).
- Thalasemia d (gangguan pembentukan rantai d)
Secara klinis talasemia dibagi dalam 2
golongan yaitu :
- Thalasemia Mayor (bentuk homozigot) Memberikan gejala klinis yang jelas
- Thalasemia Minor biasanya tidak memberikan gejala klinis
Gejala Klinis Thalasemia
Thalasemia mayor, gejala klinik telah terlihat sejak anak baru berumur kurang dari 1 tahun, yaitu:
- Lemah
- Pucat
- Perkembangan
fisik tidak sesuai dengan umur
- Berat
badan kurang
- Tidak
dapat hidup tanpa transfusi
Thalasemia
intermedia : ditandai oleh anemia mikrositik, bentuk heterozigot.
Thalasemia minor/thalasemia trait : ditandai oleh splenomegali, anemia berat, bentuk homozigot.
Thalasemia minor/thalasemia trait : ditandai oleh splenomegali, anemia berat, bentuk homozigot.
Patofisiologi Thalasemia
Penyebab anemia
pada thalasemia bersifat primer dan sekunder. Penyebab primer adalah
berkurangnya sintesis Hb A dan eritropoesis yang tidak efektif disertai
penghancuran sel-sel eritrosit intrameduler. Penyebab sekunder adalah karena
defisiensi asam folat,bertambahnya volume plasma intravaskuler yang
mengakibatkan hemodilusi, dan destruksi eritrosit oleh system
retikuloendotelial dalam limfa dan hati.
Penelitian
biomolekular menunjukkan adanya mutasi DNA pada gen sehingga produksi rantai
alfa atau beta dari hemoglobin berkurang. Tejadinya hemosiderosis merupakan
hasil kombinasi antara transfusi berulang,peningkatan absorpsi besi dalam usus
karena eritropoesis yang tidak efektif, anemia kronis serta proses hemolisis.
- Normal hemoglobin adalah terdiri dari Hb-A dengan dua
polipeptida rantai alpa dan dua rantai beta.
- Pada Beta thalasemia yaitu tidak adanya atau kurangnya rantai
Beta dalam molekul hemoglobin yang mana ada gangguan kemampuan eritrosit
membawa oksigen.
- Kelebihan pada rantai alpa pada thalasemia Beta dan Gama
ditemukan pada thalasemia alpa. Kelebihan rantai polipeptida ini mengalami
presipitasi dalam sel eritrosit. Globin intra-eritrositk yang mengalami
presipitasi, yang terjadi sebagai rantai polipeptida alpa dan beta, atau
terdiri dari hemoglobin tak stabil-badan Heinz, merusak sampul eritrosit
dan menyebabkan hemolisis.
- Reduksi dalam hemoglobin menstimulasi bone marrow memproduksi
RBC yang lebih. Dalam stimulasi yang konstan pada bone marrow, produksi
RBC diluar menjadi eritropoitik aktif. Kompensator produksi RBC terus
menerus pada suatu dasar kronik, dan dengan cepatnya destruksi RBC,
menimbulkan tidak adekuatnya sirkulasi hemoglobin. Kelebihan produksi dan
distruksi RBC menyebabkan bone marrow menjadi tipis dan mudah pecah atau
rapuh.
Pemeriksaan Penunjang
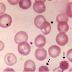
- Hasil apusan darah tepi didapatkan gambaran perubahan-perubahan
sel dara merah, yaitu mikrositosis, anisositosis, hipokromi,
poikilositosis, kadar besi dalam serum meninggi, eritrosit yang imatur,
kadar Hb dan Ht menurun.
- Elektroforesis hemoglobin: hemoglobin klien mengandung HbF dan
A2 yang tinggi, biasanya lebih dari 30 % k
THALASEMIA.Bambang Permono, IDG Ugrasena, Mia Ratwita
A
Divisi Hematologi – Bag./SMF Ilmu Kesehatan
Anak FK Unair/RSU Dr.
Thalassemia adalah
suatu kelompok anemia hemolitik kongenital herediter yang diturunkan secara
autosomal, disebabkan oleh kekurangan sintesis rantai polipeptid yang menyusun
molekul globin dalam hemoglobin.
PATOFISIOLOGI
·
Molekul globin terdiri atas
sepasang rantai-a dan sepasang
rantai lain yang menentukan jenis Hb. Pada orang normal terdapat 3 jenis Hb,
yaitu Hb A (merupakan > 96% dari Hb total, tersusun dari 2 rantai-a dan 2 rantai-b = a2b2), Hb F (<
2% = a2g2) dan HbA2 (< 3% = a2d2). Kelainan
produksi dapat terjadi pada ranta-a (a-thalassemia), rantai-b (b-thalassemia), rantai-g (g-thalassemia), rantai-d (d-thalassemia), maupun kombinasi
kelainan rantai-d dan rantai-b (db-thalassemia).
·
Pada thalassemia-b, kekurangan produksi rantai beta menyebabkan kekurangan pembentukan
a2b2 (Hb A); kelebihan rantai-a akan berikatan dengan rantai-g yang secara kompensatoir Hb F meningkat; sisanya dalam jumlah besar
diendapkan pada membran eritrosit sebagai Heinz bodies dengan akibat
eritrosit mudah rusak (ineffective erythropoesis).
DIAGNOSIS
1.
Anamnesis
Keluhan
timbul karena anemia: pucat, gangguan nafsu makan, gangguan tumbuh kembang dan
perut membesar karena pembesaran lien dan hati. Pada umumnya keluh kesah ini
mulai timbul pada usia 6 bulan.
1.
Pemeriksaan fisis
o
Pucat
o
Bentuk muka mongoloid (facies
Cooley)
o
Dapat ditemukan ikterus
o
Gangguan pertumbuhan
o
Splenomegali dan hepatomegali
yang menyebabkan perut membesar
2. Pemeriksaan penunjang
3.
Darah tepi :
·
Hb rendah dapat sampai 2-3 g%
·
Gambaran morfologi eritrosit :
mikrositik hipokromik, sel target, anisositosis berat dengan makroovalositosis,
mikrosferosit, polikromasi, basophilic stippling, benda Howell-Jolly,
poikilositosis dan sel target. Gambaran ini lebih kurang khas.
·
Retikulosit meningkat.
1.
Sumsum tulang (tidak menentukan
diagnosis) :
·
Hiperplasi sistem eritropoesis
dengan normoblas terbanyak dari jenis asidofil.
·
Granula Fe (dengan pengecatan
Prussian biru) meningkat.
1.
Pemeriksaan khusus :
·
Hb F meningkat : 20%-90% Hb
total
·
Elektroforesis Hb :
hemoglobinopati lain dan mengukur kadar Hb F.
·
Pemeriksaan pedigree:
kedua orangtua pasien thalassemia mayor merupakan trait (carrier)
dengan Hb A2 meningkat (> 3,5% dari Hb total).
1.
Pemeriksaan lain :
·
Foto Ro tulang kepala :
gambaran hair on end, korteks menipis, diploe melebar dengan trabekula
tegak lurus pada korteks.
·
Foto tulang pipih dan ujung
tulang panjang : perluasan sumsum tulang sehingga trabekula tampak jelas.
DIAGNOSIS BANDING
Thalasemia minor :
·
anemia kurang besi
·
anemia karena infeksi menahun
·
anemia pada keracunan timah
hitam (Pb)
·
anemia sideroblastik
Gejala thalasemia yang sering ditemui lesu, tidak punya
nafsu makan, lekas capai, sering terkena radang tenggorokan dan flu. Penderita
mengalami ketidakseimbangan dalam produksi hemoglobin (Hb).Penderita thalasemia tidak mampu memproduksi protein alpha/protein beta dalam jumlah yang cukup. Sehingga sel darah merahnya tidak dapat terbentuk dengan sempurna. Insiden pembawa sifat thalasemia di Indonrsia berkisar antara 6-10%, artinya dari setiap 100 orang 6-10 orang adalah pembawa sifat thalassemia.
Thalasemia diturunkan oleh orang tua yang carrier kepada anaknya. Jika ayah dan ibu menderita thalassemia maka kemungkinan anaknya untuk membawa sifat talasemia sebesar 50% .
Oleh karena pembawa sifat talassemia tidak dapat dibedakan dengan individu normal, maka statusnya hanya dapat dibedakan dengan pemeriksaan laboratorium. Pemeriksaan laboratorium untuk skrining dan diagnosis thalasemia meliputi:
- Hematologi
Rutin: untuk mengetahui kadar Hb dan ukuran sel-sel darah
- Gambaran
darah tepi : untuk melihat bentuk, warna dan kematangan sel-sel darah.
- Feritin,
SI dan TIBC : Untuk melihat status besi
- Analisis
Hemoglobin : untuk diaknosis dan menentukan jenis thalassemia.
- Analisis
DNA : untuk diaknosis prenatal (pada janin) dan penelitian.
- Pucat
- Perut
membesar, disebabkan oleh pembesaran hati dan limpa.
- Akumulasi
zat besi pada beberapa organ tubuh (kulit, jantung, paru, kelenjar
endokrin, termasuk kelenjar pertumbuhan dan kelenjar pankreas pembentuk
insulin). Mengakibatkan anak gagal tumbuh, pertumbuhan seks sekundernya
terhambat (sehingga membuatnya mandul), kulit menjadi kehitaman, tulang
rapuh, serta mudah patah.
Seperti yang telah dijelaskan di atas, thalassemia mayor dan thalassemia minor disebabkan karena kelainan genetik. Namun thalassemia mayor diturunkan dari kedua orang tua yang masing-masing memiliki satu gen thalassemia.
Penanganan yang perlu dilakukan bagi penderita thalassemia mayor adalah :
- Melakukan
transfusi darah seumur hidup.
- Mengeluarkan
zat besi yang terakumulasi akibat transfusi darah rutin secara berkala.
- Memantau fungsi
jantung, paru-paru, tulang, pankreas, maupun kelenjar endokrin lainnya.
- Transplantasi
sumsum tulang belakang untuk menjadikan penderita thalassemia mayor
menjadi thalassemia minor.
- Penanganan
secara psikososial bagi penderita thalassemia dan keluarganya.
- Melakukan
transfusi darah seumur hidup pada kondisi tertentu.
- Mengeluarkan
zat besi yang terakumulasi akibat transfusi darah rutin secara berkala.
- Penanganan
secara psikososial bagi penderita dan keluarganya.





.jpg)



2 komentar:
saya Ingin merekomendasikan dan me jelaskan bagi yg baru saja tervonis thalasemia atau yang sudah lama tervonis thalassemia coba Baca sampai habis mungkin bermanfaat.
Tegakkan atau TUNTASKAN diagnosa thalsemianya... Ini penting sekali!... Jangan hanya karena dugaan/suspect thalasemia, kemudian sudah memastikan thalasemia...
1. Sudah konfirm (pasti) hasil labnya menunjukkan thalasemia, bukan hal yg lainnya...
.
2. DAN... Ada dua kali kejadian, hbnya turun dibawah 7... (tanpa diiringi suatu serangan penyakit dari luar)... ini penting sekali, untuk menyaring thalasemia intermediate (yg tak akan bisa dipastikan dengan hasil lab saja! kecuali cek genetik!)
Maksudnya ketika terkena serangan penyakit, thaller intermediate bisa saja hbnya turun dibawah 7 dan perlu bantuan tranfusi, tapi belum tentu perlu rutin berkala!...
3. ATAU... meski Hb diatas 7, adanya salah satu saja, gejala fisik yg jelas dan menyolok, seperti terlambatnya tumbuh kembang, perubahan tulang, limpa atau liver yg membesar, jantung yg bermasalah...
Jadi jika ada (no 1 DAN no 2), ATAU (no 1 DAN no 3)Baru diputuskan masuk 'terapi tranfusi', alias masuk program tranfusi rutin berkala...
.
Sekali diputuskan masuk terapi tranfusi berkala, ATUR JARAK ANTAR TRANFUSINYA
jangan lupa terapi suportif yg lainnya (wajib bagi thalasemia mayor maupun intermediate!) yaitu jaga kecukupan asupan asam folat, vitamin B12, dan vitamin E-nya... untuk apa?
1. karena produksi darah pada thaller mayor/intermediate itu jauh lebih cepat dibanding orang normal, maka bahan dasar pembentukan sel darah, yg salah duanya yaitu asam folat dan vit B12, automatis dibutuhkan banyak itu TAK AKAN terkejar oleh asupan makanan sehari hari!... karena itulah thaller mayor/intermediate itu WAJIB konsumsi tambahan asam folat setiap harinya!...sedang untuk vit B12, karena makanan pokok kita adalah nasi, masih bisa mengandalkan asupan vitB12 dari itu...
Jika kekurangan asam folat dan vit B12, maka kondisi anemianya akan memburuk... Thalasemia PLUS defisiensi asam folat... Hbnya akan turun lebih cepat, atau bagi yg intermediate kesetimbangan hbnya akan drop (sehingga butuh bantuan tranfusi!)...
2. vitamin E adalah pilihan yg cocok untuk thaller mayor/intermediate, karena vitamin C bisa berdampak mempercepat penyerapan zat besi diusus
disamping itu, kinerja organ liver pada thaller mayor/intermediate itu jauh lebih berat dibanding orang normal... artinya .. jika thaller mayor/intermediate lalai menjaga organ livernya, maka kondisi thalasemianya akan memburuk... oleh karena itulah, vitamin liver itu penting buat thaller mayor/intermediate
bagi thaller alpha, ada yg jadi jarang tranfusinya (memang seharusnya begitu!), setelah dia konsumsi rutin asam folat, vitamin E dan vitamin liver usahakan cari yang jenis herbal jangan sintetik karna lebih baik dan aman herbal dari pada sintetik. Dan kami selama ini meme berikan vitamin itu dari dokter yusuf yang berbahan herbal tr baik dari beliau. Kami juga di sarankan oleh teman yang anaknya sudah sembuh.
Alahamdulilah selama ini kosumsi vitamin dari beliau kondisi liver dll jauh lebih baik.
Yang Ingin kosumsi vitamin tr baik seperti kami coba hubungi dokter yusuf nya langsung. Ini nomor beliau 0853-6167-52-32
Posting Komentar